What is haemophilia B?
Haemophilia B is a genetic disorder caused by missing or
defective clotting factor IX (FIX, or factor 9), causing prolonged and
often spontaneous bleeding externally and internally into the muscles
and joints.¹ Haemophilia B makes up 15–20% of all haemophilia
cases.1 Approximately 70% of the haemophilia B cases are
genetically inherited and have a positive family history for the disease.²
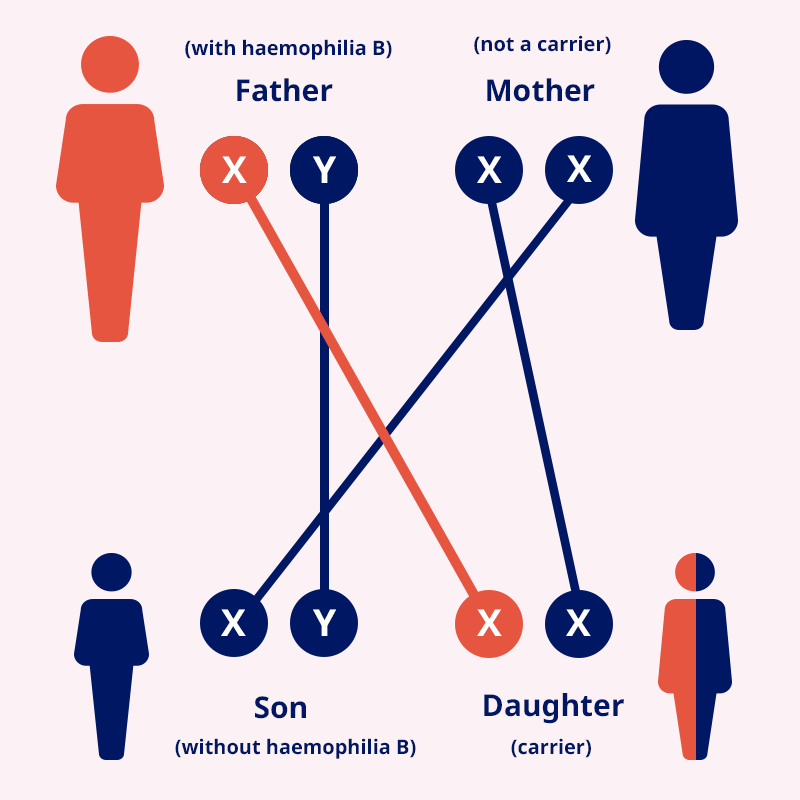
Genetics of haemophilia B
The haemophilia B gene locates on the X chromosome. Females
inherit two X chromosomes, one from their mother and one from their
father (XX). Males inherit an X chromosome from their mother and a Y
chromosome from their father (XY). So, if a son inherits an X
chromosome carrying the haemophilia B gene from his mother, he will
have haemophilia B. A father with haemophilia B cannot pass it on to
his sons but will pass the gene to his daughters.3
Because females have two X chromosomes, even if they inherit a
haemophilia B gene, they have another healthy X chromosome to
compensate. Instead, they will be carriers whose levels of the
clotting factor IX will be around half of the normal amount and may
suffer some bleeding symptoms. In that case, they will be called
symptomatic carriers. Haemophilia B can occur in females if they have
two copies of the haemophilia B gene, but it is very rare.4
Clotting factor IX
Factor IX is an essential clotting
factor required for forming normal blood clots. In response to injury,
factor IX interacts with other clotting factors to set off a chain of
reactions that form a blood clot, sealing off damaged blood
vessels.5
Normal levels of clotting factor IX vary between different people
and range from 50–150%;4 however, in people with
Haemophilia B, levels fall below 40%.6
Classification of haemophilia B according to FIX plasma level
The frequency and severity of bleeding correlate with
clotting factor IX levels as shown below.
A wide phenotypic variation is observed across patients.8
Patients with mild haemophilia typically do not have spontaneous
bleeds and are often underdiagnosed (sometimes not until
adulthood).3 Learn more about the signs, symptoms, and diagnosis of haemophilia B.
Treatment of haemophilia B
Factor IX replacement therapy
is used to prevent and treat bleeding in haemophilia B patients. The
treatment principle is simply based on replacing the person’s missing
factor IX with another factor IX used as a drug. People with severe
and some with moderate haemophilia B should take their factor IX
replacement therapy at regular intervals prophylactically to prevent
spontaneous and breakthrough bleeds.6 Research has
suggested that the higher a person’s factor IX level, the lower the
risk of spontaneous bleeds. 9 Your
physician will select the best prophylactic regimen for you based on
your bleeding phenotype, joint status, and musculoskeletal function.
Besides, your individual needs, lifestyle, and preference. 6
Types of factor IX replacement therapy
This factor IX
replacement therapy may be plasma-derived - concentrated from the
plasma or recombinant - manufactured using recombinant DNA technology.
A new generation of factor IX replacement therapy called Extended
half-life (EHL) factor IX has been developed with a longer duration of
action to allow haemophilia B patients a less frequent administration
and offer them higher factor trough levels and higher protection
against acute bleeds.6 With some EHL factor IX
products, patients with haemophilia B can stay in the
non-haemophilia range for significant amount of time with the
once-weekly dosing. 10 This is what’s
refered to by the WFH as a “
more ambitious prophylaxis ”. 6